How to tell RNA-seq library type of strand-specific for RNA-seq data (for reads mapping by Tophat)
来源:互联网 发布:alpha软件go下载 编辑:程序博客网 时间:2024/05/21 06:36
Background:
There are three library types for Tophat: fr-unstranded, fr-firststrand and fr-secondstrand. The description for these three from Tophat' documentation is list below:
Library TypeExamplesDescriptionfr-unstrandedStandard IlluminaReads from the left-most end of the fragment (in transcript coordinates) map to the transcript strand, and the right-most end maps to the opposite strand.fr-firststranddUTP, NSR, NNSRSame as above except we enforce the rule that the right-most end of the fragment (in transcript coordinates) is the first sequenced (or only sequenced for single-end reads). Equivalently, it is assumed that only the strand generated during first strand synthesis is sequenced.fr-secondstrandLigation, Standard SOLiDSame as above except we enforce the rule that the left-most end of the fragment (in transcript coordinates) is the first sequenced (or only sequenced for single-end reads). Equivalently, it is assumed that only the strand generated during second strand synthesis is sequenced.Based on my understanding,
(1) fr-unstranded is for non-strand-specific reads, and the other for strand-specific ones;
(2) fr-firststrand: for paired-end reads, the right-end of the pair is firstly sequenced (in the first round of PCR), followed by the left-end (in the second round of PCR), in other words, the first read of the fragment contains the sequence of the antisense strand and sit in the 3' end of the fragment and the second read is of the sense strand and 5' end; for single-end reads, the reads is the sequence of the sense strand (positive);
(3) fr-secondstrand: for paired-end reads, the left-end of the pair is firstly sequenced (in the first round of PCR), followed by the right-end (in the second round of PCR), in other words, the first read of the fragment contains the sequence of the sense strand and sit in the 3' end of the fragment and the second read is of the antisense strand; for single-end reads, the reads is the sequence of the antisense strand (negative);
Following graph shows the difference of the paired-end reads in these three types:
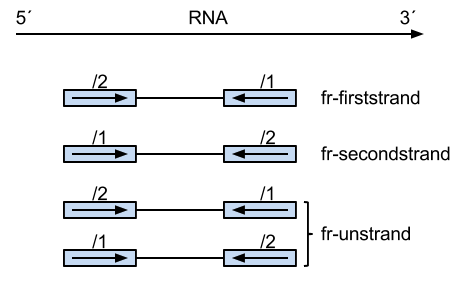 ps: "/1" means the read we get first from the fragment, and the read id, such as "seq***_1", has the same meaning.
ps: "/1" means the read we get first from the fragment, and the read id, such as "seq***_1", has the same meaning.
How to tell the library type from our law data?
Approach 1: map our reads (preprocessed read is prefered, in other words, the adapters have been removed) to the genome using UCSC genome browser with BLAT or using IGV, and tell whether the reads mapped to sense strand or antisense strand. More details can be found in: http://onetipperday.blogspot.sg/2012/07/how-to-tell-which-library-type-to-use.html,
Approach 2: map these reads with tophat using fr-firststrand and fr-second-strand respectively, and look at the file called "junctions.bed". Choose the parameter with more junctions found. More details can be found in:http://ccb.jhu.edu/software/tophat/faq.shtml#library_type
- How to tell RNA-seq library type of strand-specific for RNA-seq data (for reads mapping by Tophat)
- RNA-Seq
- RNA-Seq数据分析
- RNA-seq analysis (practice)
- RNA-Seq名词解释
- RNA-seq流程报告
- RNA-seq experiment
- RNA-seq测序数据(reads)提交NCBI
- RNA-seq与miRNA-seq联合分析
- RNA-seq 基本分析流程
- RNA-seq Data Analysis-A Practical Approach-book(2015) 笔记
- RNA-seq 流程、问题总结(一)
- RNA-Seq De novo Assembly Using Trinity
- RNA-seq差异表达分析工作流程
- 基于参考注释的RNA-seq分析
- RNA-seq Experimental Design and Quality Control
- 基于RNA-seq的基因表达分析
- RNA-Seq数据去接头(Adapter)
- UIday04
- 190 13 个球一个天平,现知道只有一个和其它的重量不同,问怎样称才能用三次就找到那 个球?
- uvaoj1339ancient cipher
- phpcms标签
- 使用Eclipse Debug调试程序详解
- How to tell RNA-seq library type of strand-specific for RNA-seq data (for reads mapping by Tophat)
- 第十五周上机项目1 “内部”寻“内幕”
- 折半查找
- 194 人人笔试
- 颜色模型
- 关于 alter table move tablespace 的一点理解
- 折半查找的递归算法
- 浅谈Hibernate里的Fetch的作用
- UVAOJ-Digit counting


